
Research
MAQC Consortium 2011–2014 (read more)
FDA SEQC, Nature Biotechnology (read more)
Bioinformatics (read more)
FEBS Journal (read more)
Gerontology (read more)
Power and Limitations of RNA-Seq: findings from the SEQC (MAQC-III) consortium
Gene activities change in response to disease and therapy and are thus of central interest in biomedical research. We have made leading contributions to a special issue of the high-impact journal Nature Biotechnology that reports a group of landmark reference studies coordinated by the U.S. Food and Drug Administration (FDA). Critical performance tests examined the precision and reproducibility of the best tools for measuring gene activities. Reproducibility forms the basis of all scientific advance, allowing others to scrutinize claims and combine results. Beyond basic and applied research, reproducibility is also the foundation for any effective development of new therapies and interventions. In particular, regulators like the FDA, the EMA, and CFDA rely on reproducible research to approve proposed novel drugs and therapies, including the latest precision medicine. Consequently, the FDA has brought together over 150 researchers from 12 countries in the Sequencing Quality Consortium (SEQC) to test the latest tools for measuring gene activity and compare results across different laboratories and technologies.
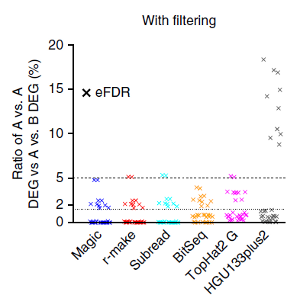
With our group taking a leading role in the consortium, we have determined new guidelines that can help scientists and clinicians get reliable analysis results, making them comparable across different laboratories and technologies. Our studies covered methods for assessing gene activities by RNA-Seq, using the latest next-generation sequencing platforms. Comparisons show their performances relative to already established methods like quantitative PCR and microarrays. For all methods, traditional analyses looking for differences in gene expression identify thousands of false positives, as reflected in comparisons of identical samples. For microarrays, these false positives can be controlled by extra filters for effect size. In addition, RNA-seq also requires filters for expression strength due to the high sampling fluctuations at lower read counts. The figure above plots estimates of the empirical False Discovery Rate (eFDR). For both RNA-Seq (colours) and microarrays (grey), the application of appropriate filters in general improved the eFDR to excellent values below 1.5% (second dashed line).
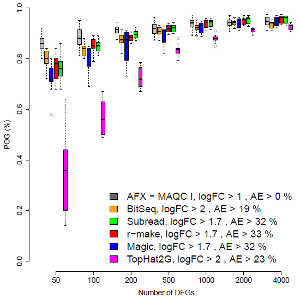
Most screens for genes implicated in a disease (or any other phenotype of interest) generate ranked lists of candidates. Their reproducibility is therefore of direct immediate interest. The figure on the right plots the percentage agreement across different laboratory sites. On the left, the top ranked 50 genes are compared. Agreements for larger lists are shown further to the right. In general, the highest ranked genes are of particular interest. For most RNA-Seq methods (colours), the application of appropriate filters yielded good reproducibility, comparable to that of microarrays (grey). One RNA-Seq method, however, performed poorly, despite being popular and applied in many past and present studies.
Experimental and computational work by our group has made key contributions to establishing protocols for next-generation sequencing based tools to work reliably across laboratories. Built-in controls in the study design further allowed an assessment of accuracy and information content of gene expression measurements by the different technological platforms. Tools based on next-generation sequencing showed different strengths compared to microarray platforms, with the competing technologies complementing one another, showing promising paths to future improvements of gene expression profiling methods.
Moreover, our work has raised a number of exciting questions. For instance, we found many thousands of new gene assembly junctions validated by independent measurements. Do they have a function? And what do they do? Further studies will be required to shed more light on the potential value of structural gene variant discoveries by next-generation sequencing.
Paweł P. Łabaj, Peter Sykacek, Nancy Stralis Pavese, David P. Kreil

Read more
Su, Z;1 Łabaj, PP;1 Li, S;1 … Sykacek, P; … Stralis-Paves, N; … Tong, W; Kreil, DP;* Mason, CE;* and Shi, L.* A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nature Biotechnology, 2014; Manuscript and analysis lead.
Li, S;1 Łabaj, PP;1 Zumbo, P;1 Sykacek, P; … Kreil, DP;* Mason, CE.* Detecting and correcting systematic variation in large-scale RNA sequencing data. Nature Biotechnology, 2014;
Munro, SA; … Kreil, DP; Łabaj, PP; … Stralis-Pavese, N; … Salit, M. Assessing technical performance in differential gene expression experiments with external spike-in RNA control ratio mixtures (ArXiv Preprint). Nature Communications, 2014; accepted
Wang, C; … Łabaj, PP; Kreil, DP; … Tong, W. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nature Biotechnology, 2014;
Hu, J; Su, Z; Hong, H; Thierry-Mieg, J; Thierry-Mieg, D; Kreil, DP; Mason, CE; Tong, W; Shi, L. Cross-platform ultradeep transcriptomic profiling of human reference RNA samples by RNA-Seq. Scientific Data, 2014; 1; 140020
1 joint first authorship
* joint senior/corresponding authorship